- Стресс испытания лекарственных препаратов
- Сроки годности лекарственных средств (ОФС.1.1.0009.15)
- Основные термины и определения
- Общие положения
- Долгосрочные испытания стабильности лекарственных средств
- Испытания стабильности методом «ускоренного старения»
- Испытания стабильности методом экстраполяции
- Испытания стабильности методом крайних вариантов
- Испытания стабильности матричным методом
- Исследование влияния упаковки на стабильность лекарственного средства
- Исследования стабильности лекарственных препаратов после восстановления или разведения
- Стресс-исследования и фотостабильность
Стресс испытания лекарственных препаратов

Профилактика и лечение сосудистых заболеваний головного мозга является в настоящее время важной и актуальной задачей во всем мире. Инсульт – это серьезная проблема общественного здравоохранения, поскольку ежегодно около 15 миллионов человек во всем мире переносят инсульт [1; 2]. В нашей стране инсульт поражает более 450 000 человек в год, заболеваемость и смертность от острых нарушений мозгового кровообращения находится на 2-м месте среди других заболеваний. Для того чтобы избежать высокой смертности и инвалидности, необходимо раннее выявление и адекватное лечение инсульта, а также обязательное проведение реабилитационных мероприятий, которые препятствуют возникновению рецидива. Одним из компонентов реабилитации является медикаментозная терапия с использованием церебропротекторных, ноотропных и стресс-протекторных препаратов. Одним из перспективных направлений создания новых препаратов, обладающих церебропротекторным действием, является разработка инновационных соединений на основе пептидных биорегуляторов.
Новое потенциальное лекарственное средство (ЛС) на основе пептидэргического нейро- и стресс-протектора Nα-ацетил-D-лизил-лизил-аргинил-аргинил-амид является кандидатным препаратом для лечения нарушений мозгового кровообращения. Проведенные ранее доклинические исследования специфической фармакологической активности потенциального ЛС показали его высокую нейропротекторную эффективность in vivo [3]. Было показано, что данное потенциальное ЛС обладает антиишемической, антигипоксической, ноотропной и актопротекторной активностями [4]. Выявлено, что Nα-ацетил-D-лизил-лизил-аргинил-аргинил-амид, являющий активным фармакологическим ингредиентом потенциального ЛС, оказывает интегральное нейропротекторное действие, нормализует уровень провоспалительных цитокинов IL-1β и TNF-α, а также повышает содержание противовоспалительного интерлейкина-4 в головном мозге. Защитное действие пептида при церебральной ишемии характеризуется также нейротрофическим компонентом.
Целью данной работы являлись доклинические исследования острой и хронической токсичности потенциального ЛС «Лизаргам» на основе пептидэргического нейро- и стресс-протектора для лечения нарушений мозгового кровообращения.
Материалы и методы исследования
Исследуемое потенциальное ЛС «Лизаргам, спрей назальный дозированный 0,5% и 1,0%» (далее потенциальное ЛС «Лизаргам»), один флакон потенциального ЛС содержит активный фармацевтический ингредиент («D-лизаргам, ацетат») – 15 (0,5%) или 30 (1,0%) мг; физиологический раствор (ФР) — 3,0 мл; D-маннит — 30 мг. «D-лизаргам, ацетат» представляет собой синтетический тетрапептид последовательности Nα-ацетил-D-лизил-лизил-аргинил-аргинил-амид в виде ацетатной соли. Терапевтическая доза потенциального ЛС у человека составляет 20 мкг/кг. Эта доза рассчитывалась исходя из данных по изучению специфической активности потенциального ЛС на экспериментальных животных и коэффициента экстраполяции доз мышей и крыс на человека в соответствии с руководством Т.А. Гуськовой [9].
Доклинические исследования были выполнены на основе рекомендаций действующих методических документов 6. Животные поступили из питомника РАМН «Рапполово» Ленинградской области. Основные правила содержания и ухода соответствовали правилам, принятым Европейской конвенцией по защите позвоночных животных (Страсбург, 1986 г.). Объем работ и перечень процедур был одобрен биоэтической комиссией института. Исследования проводили на 3 видах животных: белых беспородных крысах обоего пола (масса 180-200 грамм, возраст 9–10 недель), белых беспородных мышах обоего пола (19–21 г, 9–10 недель) и на кроликах породы шиншилла обоего пола (2-2,5 кг, 2-3 месяца).
Острая токсичность потенциального ЛС была изучена на мышах, крысах и кроликах. Период наблюдения после введения возрастающих доз составил 14 дней, для достижения высоких доз потенциальное ЛС вводили многократно в течение 10 часов. Эксперименты были проведены при интраназальном (и/н) и внутривенном (в/в) введении потенциального ЛС по одинаковым схемам. Мыши и крысы были случайным образом разделены на группы (по 5 самок и 5 самцов). В опытных группах мышам и крысам и/н вводили потенциальное ЛС в дозах 2, 20, 50 мг/кг; в/в — в дозах 20, 200, 500 мг/кг. И/н потенциальное ЛС вводилось животным при помощи автоматической одноканальной пипетки 10-100 мкл с соответствующим наконечником, введение осуществлялось в каждый носовой ход. В/в введение производилось в соответствии с требованиями [6], в максимальной дозе 500 мг/кг (50 мл/кг) каждой крысе вводили не более 2 мл за раз с интервалом 2 часа в течение 10 часов. Кролики были случайным образом разделены на группы (по 3 самки и по 3 самца в каждой). Кроликам в опытной группе потенциальное ЛС вводили и/н в дозе 25 мг/кг, в/в — в дозе 400 мг/кг. В контрольных группах животным по тем же схемам вводили ФР.
Во всех экспериментах проводили наблюдение за лабораторными животными в течение 14 суток после введения потенциального ЛС. Проводили клинический осмотр (ежедневно) до начала эксперимента (фоновые значения), а также выполняли взвешивание на 2, 7 и 14-й день после введения. У крыс и кроликов измеряли частоту сердечных сокращений (ЧСС) и частоту дыхательных движений (ЧДД). На 14-е сутки все животные были подвергнуты эвтаназии и патоморфологическому исследованию.
Оценка местнораздражающего действия была проведена на 3 кроликах-самцах. На слизистую оболочку правого глаза наносили 0,1 мл 1% потенциального ЛС, на слизистую оболочку левого глаза — 0,1 мл ФР. Наблюдение проводили в течение 24 часов с момента нанесения потенциального ЛС.
Испытания на пирогенность были проведены на кроликах-самцах и соответствовали требованиям ГФ XIII, ОФС 1.2.4.0005.15 [8]. Потенциальное ЛС вводили в ушную вену в объеме 0,2 мл/кг. Температуру измеряли 2 раза до начала исследования и каждые 30 минут в течение 3 часов после введения потенциального ЛС.
Хроническая токсичность потенциального ЛС была изучена на крысах, случайным образом разделенных на 4 группы (по 15 самок и 15 самцов в каждой). Потенциальное ЛС вводили и/н ежедневно, в течение 90 дней в 3 дозах: 1 мг/кг (50-кратная терапевтическая доза для человека, минимально возможная для экспериментального введения крысам), 3 и 5 мг/кг. Хроническая токсичность потенциального ЛС была также изучена на кроликах, самцах и самках, которые были случайным образом разделены на группы (по 3 самки и 3 самца в каждой). Потенциальное ЛС вводили и/н ежедневно, в течение 90 дней в 3 дозах: терапевтической дозе для кролика — 0,06 мг/кг (доза рассчитана путем умножения терапевтической дозы для человека на коэффициент пересчета по площади поверхности тела кролика 3,2); 3 мг/кг и максимальной — 5 мг/кг (более чем в 250 раз превышающей терапевтическую дозу для человека). Крысам и кроликам в контрольных группах по той же схеме вводили ФР.
Клинический осмотр животных выполняли ежедневно, в течение эксперимента регистрировали интегральные показатели состояния организма всех животных, а также патоморфологическое и патогистологическое исследования по окончании эксперимента. Физиологические исследования проводили до начала исследования, а также на 30-й и на 90-й день после начала введения потенциального ЛС.
Офтальмологическое исследование проводили на тех же сроках, оценивая состояние слизистых оболочек, наличие правильных роговичных рефлексов и измеряя величину зрачка и ширину глазной щели. Для лабораторных исследований собирали образцы крови и костный мозг.
Клинический анализ крови выполняли на автоматическом гематологическом анализаторе Abacus Junior vet5 (Diatron, Венгрия) и на мазках крови, окрашенных по Романовскому-Гимзе. Биохимические показатели в сыворотках крови (аланинаминотрансфераза (АлАТ), аспартатаминотранфераза (АсАТ), билирубин, глюкоза, креатинин, мочевина, общий белок, холестерин, щелочная фосфатаза) оценивали на полуавтоматическом биохимическом анализаторе Chem-7 (Erba, Чехия). Исследование мочи выполняли на экспресс-анализаторе мочи DocUReader (ANALYTICON Biotechnologies AG, Германия). Кроме того, проводили микроскопический анализ миелограммы на мазках смывов костного мозга и подсчет количества клеток костного мозга. Все тесты выполнялись по стандартным протоколам и инструкциям производителей наборов.
На основании результатов эксперимента по изучению хронической токсичности рассчитывали расчетный безопасный курс (РБК) и индекс безопасности (ИБ) потенциального ЛС при клиническом применении в соответствии с инструкцией Т.А. Гуськовой [9].
Патоморфологическое исследование после завершения экспериментов включало в себя некропсию, макроскопическое исследование и взвешивание внутренних органов. Также рассчитывали отношение массы органа к массе тела.
Патогистологическое исследование было выполнено с использованием образцов тканей, полученных от животных, которым вводили потенциальное ЛС в максимальной дозе, а также образцов тканей от животных контрольных групп. Материал фиксировали в 10%-ном формалине, обезвоживали, заключали в парафин и изготовляли гистологические срезы, которые окрашивали гематоксилином и эозином («Биовитрум», Россия). Препараты изучали в световом микроскопе DMLB (Leica Microsystems AG, Германия).
Статистический анализ проводили с помощью программы Microsoft Excel 2007 (Microsoft Corporation). Подсчитывали средние значения и ошибки среднего (М±m). Сравнение показателей между группами проводили с помощью непарного Т-критерия Стьюдента с неравными отклонениями, а также по U-критерию Манна-Уитни. Отличия считали достоверными при р 5) потенциальное ЛС «Лизаргам, спрей назальный» относится к III классу малотоксичных лекарственных препаратов.
- Изучение хронической токсичности на кроликах
Ежедневный клинический осмотр кроликов всех экспериментальных групп не выявил каких-либо значимых различий между группами, гибели подопытных животных не наблюдалось. Измерение массы тела показало, что данный параметр равномерно увеличивался на протяжении всего срока исследования как в контрольной, так и во всех опытных группах. Анализ показателей массы тела, потребления корма и воды, а также физиологических параметров (ЧДД, ЭКГ и ЧСС и АД) показал, что достоверные различия между всеми экспериментальными группами отсутствуют. Офтальмологическое исследование не выявило различий между экспериментальными группами, получавшими потенциальное ЛС или ФР.
Представленные в таблице 6 данные подтверждают, что ежедневное и/н введение потенциального ЛС в дозах 0,06-5 мг/кг в течение 30 и 90 дней не оказывало значительного влияния на количественный и качественный состав периферической крови кроликов. Также не было выявлено влияния потенциального ЛС на состояние красного костного мозга, биохимический состав крови, мочи и функциональную активность почек.
Оценка влияния ежедневного интраназального введения потенциального лекарственного средства «Лизаргам» на показатели периферической крови кроликов-самцов
Источник
Сроки годности лекарственных средств (ОФС.1.1.0009.15)
» data-shape=»round» data-use-links data-color-scheme=»normal» data-direction=»horizontal» data-services=»messenger,vkontakte,facebook,odnoklassniki,telegram,twitter,viber,whatsapp,moimir,lj,blogger»>
Сроки годности лекарственных средств (ОФС.1.1.0009.15)
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
Взамен ГФ ХII, ч.1, ОФС 42-0075-07
Требования данной статьи распространяются на методы определения стабильности лекарственных средств промышленного производства, которые лежат в основе установления их сроков годности.
Основные термины и определения
Срок годности – период времени, в течение которого лекарственное средство полностью отвечает всем требованиям нормативной документации, в соответствии с которой оно было произведено и хранилось.
Стабильность — способность лекарственного средства сохранять химические, физические, микробиологические, биофармацевтические и фармакологические свойства в определенных границах на протяжении срока годности.
Долгосрочные испытания стабильности – испытания, проводимые в соответствии с заявленными в нормативной документации условиями хранения лекарственного средства с целью установления или подтверждения срока годности.
Испытания стабильности методом «ускоренного старения» – испытания, проводимые при повышенной температуре с целью установления или подтверждения срока годности лекарственного средства.
Стресс-исследования – испытания стабильности в стресс-условиях, проводимые с целью исследования вынужденного процесса разложения (установления продуктов и механизмов разложения) лекарственного средства.
Матричный метод исследования стабильности (matrixing) – метод исследования, при котором в определенный момент времени исследуется лишь подгруппа из общего числа образцов всех комбинаций факторов, подлежащих изучению.
Метод крайних вариантов (bracketing) – метод изучения стабильности, при котором во всех временных точках по полному протоколу тестируют только образцы с крайними вариантами факторов.
Экстраполяция − способ получения информации о будущих данных на основании имеющихся.
Дата производства лекарственных препаратов – дата выполнения первой производственной операции, связанной со смешиванием фармацевтической субстанции с другими компонентами. Для фармацевтических субстанций датой производства считается начальная дата операции по фасовке и упаковке.
Дата выпуска – дата поступления или разрешения поступления лекарственного средства в обращение.
Общие положения
Срок годности лекарственного средства устанавливается экспериментально при хранении в течение определенного времени в условиях и упаковке, регламентируемых нормативной документацией, и по мере накопления данных он может быть изменен как в сторону увеличения, так и в сторону уменьшения.
В основу определения сроков годности положено изучение стабильности лекарственного средства с использованием химических и физико-химических методов анализа, указанных в общих фармакопейных статьях, а также, в случае необходимости, других специальных методов исследований, например, биологических методов анализа, фармакологических испытаний.
Срок годности лекарственных препаратов устанавливают независимо от сроков годности фармацевтических субстанций. Однако для лекарственных препаратов, произведенных путем фасовки фармацевтических субстанций, следует учитывать, что стабильность лекарственных препаратов может зависеть от остаточного срока годности используемой фармацевтической субстанции.
Под остаточным сроком годности подразумевается период времени, оставшийся до окончания установленного срока годности лекарственного средства.
Также необходимо учитывать и оценивать влияние на стабильность лекарственных препаратов фактора длительного хранения нерасфасованной и промежуточной продукции до передачи с одного производственного участка на другой или на участок упаковки.
Первоначальный срок годности лекарственного средства определяет производитель (разработчик) лекарственного средства при подготовке проекта нормативной документации.
После регистрации лекарственного средства и начала промышленного выпуска производитель (разработчик) обязан продолжить работы по изучению стабильности лекарственного средства с целью подтверждения или уточнения его срока годности.
Для лекарственных препаратов не рекомендуется устанавливать сроки годности более 5 лет, даже если результаты изучения стабильности позволяют это сделать.
Изменение сроков годности лекарственных средств или условий хранения утверждается в установленном порядке на основе данных, подтверждающих обоснованность заявленных изменений.
Отсчет срока годности промышленной серии лекарственного средства проводят от даты выдачи разрешения на ее реализацию (даты выпуска). При нормальных обстоятельствах период до даты выдачи такого разрешения не должен превышать 30 суток от даты производства.
Если разрешение на реализацию серии лекарственного средства выдано по истечении установленных 30 суток от даты производства, то началом отчета срока годности лекарственного средства следует считать дату производства.
Данный подход не применим для иммунобиологических лекарственных препаратов, таких как вакцины, сыворотки, анатоксины и аллергены, для лекарственных препаратов, полученных из крови и плазмы крови человека и животных, а также для лекарственных препаратов, полученных с использованием биотехнологических процессов и методов.
На основании изучения свойств лекарственного средства устанавливают оптимальные требования к первичной и вторичной упаковке и условиям хранения.
После установления оптимальных требований к первичной и вторичной упаковке и условиям хранения производитель (разработчик) лекарственного средства экспериментально определяет сроки годности лекарственного средства, осуществляя его хранение в рекомендованной упаковке и в указанных условиях с целью обнаружения скрытых факторов, которые могут повлиять на устойчивость лекарственного средства при хранении. Для этого от каждой из не менее чем 3 серий образца лекарственного средства, специально произведенных по условиям лабораторного или опытно-промышленного регламента, отбирают и упаковывают часть из них в количестве, достаточном для исследования стабильности лекарственного средства.
При изучении стабильности лекарственных средств, расфасованных в крупногабаритную первичную упаковку (фляги, бутыли, железные бочки, жестяные барабаны, пакеты полимерные, мешки бумажные трех- и четырехслойные и т.д.), допускается использование аналогичной упаковки меньшей емкости, достаточной для моделирования условий оригинальной упаковки.
Перед началом испытания проводят анализ лекарственного средства по всем показателям, предусмотренным проектом нормативной документации. Испытания по показателям, которые не изменяются в процессе хранения, а также испытания по показателям, изменения которых в процессе хранения не происходят в сторону ухудшения качества лекарственного средства, допускается не включать в протоколы исследования стабильности лекарственного средства.
На основании полученных результатов производитель (разработчик), изучающий стабильность лекарственного средства, определяет срок годности с указанием вида упаковки, требуемых условий хранения и транспортирования и вносит эти данные в проект нормативной документации.
При исследовании стабильности лекарственного препарата одновременно с изучением стабильности действующего и вспомогательного веществ оценивают их совместимость.
Дизайн программы испытаний на стабильность должен учитывать климатические условия в той области, где планируется использование лекарственных средств.
В табл. 1 приведены средние значения температуры и влажности, установленные в соответствующих климатических зонах.
Таблица 1. Средние значения температуры и влажности в климатических зонах
| Наименование климатической зоны | Температура, ° С | Относительная влажность, % |
| Зона I – умеренный климат | 21 | 45 |
| Зона II – субтропический климат с возможной высокой влажностью | 25 | 60 |
| Зона III – жаркий и сухой климат | 30 | 35 |
| Зона IVА – жаркий и влажный климат | 30 | 65 |
| Зона IVБ – жаркий и очень влажный климат | 30 | 75 |
Примечание. Климатические зоны — разделение планеты на 4 зоны, основанное на преобладающих годовых климатических условиях.
Рекомендуется проводить изучение стабильности лекарственных препаратов, произведенных из разных серий фармацевтической субстанции.
Для некоторых лекарственных препаратов в таких лекарственных формах, как растворы, суспензии, эмульсии и др., при необходимости проводят исследования по изучению влияния на их стабильность отрицательных температур (циклы замораживания и оттаивания).
Долгосрочные испытания стабильности лекарственных средств
Долгосрочные испытания должны проводиться в рекомендованной для данного лекарственного средства первичной и вторичной упаковке при постоянной верхней (наиболее высокой) температуре установленного режима хранения в течение всего заявленного срока годности.
В ряде случаев могут требоваться дополнительные испытания при нижней температуре установленного режима хранения (например, для мягких лекарственных форм, для которых возможны изменения их физико-химического состояния при пониженных температурах).
Образцы лекарственных средств, находящиеся на изучении стабильности, подлежат проверке по показателям качества нормативной документации в следующие сроки:
- в течение первого года хранения – через каждые 3 мес;
- в течение второго и третьего года хранения – через каждые 6 мес;
- после третьего года хранения – через каждые 12 мес.
Испытания стабильности методом «ускоренного старения»
Метод «ускоренного старения» преимущественно используется для определения сроков годности фармацевтических субстанций, представляющих собой вещества с установленным химическим строением, и лекарственных препаратов, содержащих эти вещества в качестве действующих.
Не рекомендуется использовать этот метод определения для лекарственного растительного сырья, лекарственных растительных препаратов, гомеопатических лекарственных средств, термолабильных фармацевтических субстанций и лекарственных препаратов, иммунобиологических лекарственных препаратов, препаратов крови и др.
Срок годности, установленный с помощью метода «ускоренного старения», не должен превышать 3 лет для антибиотиков, полученных микробиологическим или полусинтетическим путем, и их лекарственных форм – 2 лет. Метод не применим для увеличения ранее установленного срока годности лекарственного средства свыше 3 лет.
Метод «ускоренного старения» заключается в выдерживании испытуемого лекарственного средства при температурах и влажности, превышающих температуру и влажность его хранения в процессе обращения.
В данной ОФС приводятся рекомендации по изучению сроков годности лекарственных средств методом «ускоренного старения» с использованием измененного температурного режима. При повышенных температурах, как правило, ускоряются протекающие в лекарственных средствах физико-химические процессы, приводящие со временем к нежелательным изменениям качества. Таким образом, при повышенной температуре промежуток времени, в течение которого контролируемые показатели качества лекарственного средства сохраняются в допустимых пределах (экспериментальный срок годности), искусственно сокращается в сравнении со сроком годности при температуре хранения. Это позволяет значительно сократить время, необходимое для установления срока годности.
По результатам, полученным в процессе «ускоренного старения» лекарственного средства, можно решить также обратную задачу, т.е. установить температуру хранения, обеспечивающую какой-либо заданный срок годности.
Срок годности (С) при температуре хранения (tхр). связан с экспериментальным сроком годности (СЭ) при повышенной температуре экспериментального хранения (tэ) следующей зависимостью:
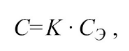
где коэффициент соответствия (К)
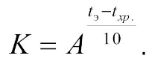
Температурный коэффициент скорости химической реакции (A) принят равным 2,5.
Примечания
- Приведенная зависимость основана на правиле Вант-Гоффа о 2-4-кратном росте скоростей химических реакций при увеличении температуры на 10°С.
- В отдельных случаях возможно использование экспериментально определенных уточненных значений коэффициента A, а также прогнозирования сроков годности на основании более строгих зависимостей, например уравнения Аррениуса.
В табл. 2 приведены значения коэффициентов соответствия K для различных значений разности температур экспериментального и обычного хранения при A = 2,5.
Таблица 2. Значения коэффициентов соответствия (K) в зависимости от температурного интервала
| № | (tэ—tхр), о С | K |
| 1 | 10 | 2,5 |
| 2 | 15 | 4,0 |
| 3 | 20 | 6,3 |
| 4 | 25 | 9,9 |
| 5 | 30 | 15,6 |
| 6 | 35 | 24,7 |
Примечание. Условные обозначения: К ‑ коэффициент соответствия; tэ ‑ температура экспериментального хранения; tхр ‑ температура обычного хранения.
Для опытов по «ускоренному старению» лекарственных средств должны использоваться термостаты, термошкафы, климатические камеры или другие устройства, позволяющие автоматически поддерживать заданную температуру экспериментального хранения tэ в течение всего опыта с точностью ±2 °С.
Наиболее высокая температура экспериментального хранения должна обеспечивать получение результатов, необходимых для оценки сроков годности, в кратчайшие промежутки времени. Однако эта температура не должна превышать пределов, за которыми происходят изменения агрегатного состояния лекарственного средства или разрушение упаковочного материала.
Рекомендуются следующие предельные температуры экспериментального хранения:
| — для индивидуальных веществ | + 60 °С |
| — для парентеральных растворов в стеклянной упаковке, таблеток, капсул | + 60 °С |
| — для парентеральных растворов в полимерной упаковке, мазей, линиментов | + 40 °С |
| — для суппозиториев и аэрозолей: | + 30 °С |
Воздействие света на испытуемые образцы должно быть исключено.
Не рекомендуется установление срока годности методом «ускоренного старения» для эмульсий.
Определение сроков годности методом «ускоренного старения» должно проводиться не менее чем на 3 сериях лекарственного средства.
Температура экспериментального хранения (tэ) должна превышать температуру хранения (tхр) не менее чем на 10 °C.
Наблюдение за качеством изучаемых образцов лекарственного средства должно проводиться по показателям, предусмотренным нормативной документацией, с учетом общих положений настоящей статьи.
Показатели качества лекарственного средства в процессе «ускоренного старения» определяют через промежутки времени, эквивалентные 6 месяцам хранения при условиях хранения, указанных в проекте нормативной документации.
Количество образцов лекарственного средства, предназначенных для экспериментального хранения, должно быть достаточным для проведения исследований, предусмотренных планом эксперимента.
Началом экспериментального хранения считается момент помещения лекарственного средства в термостатирующее устройство, а концом – либо момент, когда истекает экспериментальный срок хранения, соответствующий не менее чем двухлетнему сроку годности, либо момент, когда показатели качества лекарственного средства перестают удовлетворять требованиям нормативной документации.
Сроки экспериментального хранения при различных температурах представлены в табл. 3.
Таблица 3. Сроки экспериментального хранения в зависимости от температурного интервала
| № | Срок годности | Сроки экспериментального хранения, сут | |
| 1 | 2 года | 10 | 292 |
| 15 | 182 | ||
| 20 | 116 | ||
| 25 | 74 | ||
| 30 | 47 | ||
| 35 | 30 | ||
| 2 | 3 года | 10 | 438 |
| 15 | 274 | ||
| 20 | 174 | ||
| 25 | 111 | ||
| 30 | 71 | ||
| 35 | 45 | ||
| 3 | 4 года* | 10 | 584 |
| 15 | 365 | ||
| 20 | 232 | ||
| 25 | 148 | ||
| 30 | 94 | ||
| 35 | 60 | ||
| 4 | 5 лет* | 10 | 730 |
| 15 | 457 | ||
| 20 | 290 | ||
| 25 | 185 | ||
| 30 | 117 | ||
| 35 | 74 |
Примечание
* — в случае подтверждения срока годности, равного ранее утвержденному.
Условные обозначения: tэ — температура экспериментального хранения; tхр — температура обычного хранения.
Для вычисления срока годности экспериментальный срок годности, выраженный в сутках (или часах), умножают на коэффициент соответствия K (см. табл. 1).
Если промежуток времени Со между датой производства/изготовления лекарственного средства и началом его экспериментального хранения превышает 30 сут (но не более 90 сут), и оно в это время хранилось в обычных условиях, расчет срока годности С проводят по уравнению:
Если сроки годности, установленные на различных сериях лекарственных средств, отличаются друг от друга, за срок годности принимают минимальное из полученных значений.
При необходимости температуру tхр, позволяющую обеспечить заданный срок годности С, рассчитывают по формуле:
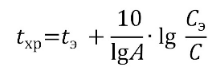
Данные, полученные с использованием метода «ускоренного старения», должны быть подкреплены обязательствами предприятия (разработчика) по продолжению изучения стабильности в условиях долгосрочных испытаний в течение всего заявленного срока годности.
Испытания стабильности методом экстраполяции
При достаточном обосновании допускается экстраполяция данных, полученных по результатам долгосрочного хранения.
Экстраполяцию проводят с помощью статистической обработки данных. Если полученные результаты свидетельствуют о незначительной деградации и малой вариации, статистический анализ может не проводиться.
Методом экстраполяции предлагаемый срок годности может быть увеличен не более чем в 2 раза, но не более чем на 12 мес, по сравнению с долгосрочными испытаниями.
Данные, полученные с использованием метода экстраполяции, должны быть подкреплены обязательствами предприятия (разработчика) по продолжению изучения стабильности в условиях долгосрочных испытаний в течение всего заявленного срока годности.
Испытания стабильности методом крайних вариантов
При изучении стабильности лекарственных средств допускается проведение исследования методом крайних вариантов.
При использовании метода крайних вариантов во всех временных точках по полному протоколу тестируют только образцы с крайними (предельными) вариантами факторов (например, дозировки, размер упаковки (тары) и (или) номинальный объем). Такой протокол предполагает, что стабильность любых промежуточных вариантов соответствует стабильности исследуемых крайних вариантов.
Исследование крайних вариантов допускают в отношении нескольких дозировок с пропорциональным составом; в случае одного и того же вида упаковки, если при прочих равных условиях имеются различия в размере упаковки или номинальном объеме лекарственного препарата.
Испытания стабильности матричным методом
При использовании матричного метода в определенный момент времени исследуется лишь подгруппа из общего числа образцов всех комбинаций факторов, подлежащих изучению. В очередной момент времени проводят исследование другой подгруппы образцов всех комбинаций факторов. К различным факторам одного и того же лекарственного препарата относят, например, совокупность различных серий, различных дозировок, различных размеров одной и той же укупорочной системы упаковки и, в ряде случаев, различных укупорочных систем упаковки.
Применение матричного метода допускают в отношении нескольких дозировок, идентичных или близких по составам.
Исследование влияния упаковки на стабильность лекарственного средства
Изменения качества лекарственного препарата могут быть вызваны взаимодействием лекарственного средства и системы упаковки, включающей укупорочные средства. Если для жидких лекарственных препаратов (кроме тех, что находятся в запаянных ампулах) нельзя исключить отсутствие взаимодействия, то в испытания на стабильность включают образцы в перевернутом или горизонтальном положениях (т.е. образцы, которые контактируют с укупорочным средством, например, пробкой), наряду с вертикально установленными образцами, для определения влияния материала укупорочного средства (пробки) на качество лекарственного препарата. Результаты экспериментальных исследований должны фиксировать все сочетания различных систем упаковки (укупорки), анализируемых лекарственных средств.
Для лекарственных средств в многодозовой упаковке, кроме стандартных данных, необходимых для традиционной упаковки одноразового использования (например, флакона), заявитель должен провести испытания, подтверждающие способность упаковки выдержать условия повторного открывания/закрывания и при этом сохранить качество и эффективность лекарственного средства на протяжении всего срока применения.
К основным факторам, оказывающим влияние на лекарственный препарат после вскрытия упаковки, относятся микробное загрязнение и физико-химическая деградация.
В исследование стабильности лекарственных препаратов после вскрытия первичной упаковки следует включать не менее 2 серий; при этом, по крайней мере, одна серия должна быть с истекающим сроком годности.
Проверку показателей на соответствие требованиям нормативной документации осуществляют в первую временную точку, как минимум одну промежуточную, а также в последнюю временную точку предлагаемого срока годности вскрытого лекарственного препарата.
Анализ лекарственного препарата проводят по всем показателям нормативной документации, которые могут меняться в процессе хранения (за исключением показателей, изменения по которым в процессе хранения не могут происходить в сторону ухудшения качества), и обязательно должны включать контроль на микробиологическую чистоту или стерильность.
Исследования стабильности лекарственных препаратов после восстановления или разведения
Если предполагается возможность хранения восстановленного твердого лекарственного препарата или разведенного концентрированного лекарственного препарата в течение определенного периода времени, должны проводиться исследования стабильности приготовленного таким образом препарата.
Цель изучения стабильности восстановленных препаратов – определить срок, в течение которого после восстановления или разведения лекарственного препарата его качество продолжит соответствовать требованиям нормативной документации, и лекарственный препарат может применяться по назначению.
Изучению стабильности подлежат восстановленные лекарственные препараты, приготовленные с использованием всех возможных для растворения/разведения лекарственных препаратов растворителей, указанных в инструкции по медицинскому применению.
Условия хранения восстановленного лекарственного препарата могут отличаться от условий хранения исходного лекарственного препарата.
Для подтверждения стабильности восстановленного лекарственного препарата допускается предоставлять данные, полученные для 2 серий, при этом, по крайней мере, одна серия должна быть с истекающим сроком годности.
Проверку показателей на соответствие требованиям нормативной документации рекомендуется осуществлять в первую и последнюю временные точки предлагаемого срока годности восстановленного лекарственного препарата.
Анализ лекарственного препарата проводят по всем показателям, которые могут меняться в процессе хранения, и обязательно должен включать контроль на стерильность или микробиологическую чистоту.
Стресс-исследования и фотостабильность
Помимо установления срока годности и выбора условий хранения изучение стабильности оригинальных лекарственных препаратов и фармацевтических субстанций проводится с целью установления наиболее вредного влияния внешних факторов (высокие или низкие температуры, влага, кислород и другие компоненты воздуха, свет и т.п.) в зависимости от времени и условий их воздействия.
Стресс-исследования допускается проводить на одной серии лекарственного средства.
Неотъемлемой частью стресс-исследований является исследование фотостабильности.
Объем исследований лекарственного средства должен определяться на основании наличия или отсутствия изменений, возникших в результате влияния света.
Условия хранения лекарственных средств, указанные в нормативной документации, должны соблюдаться на всех этапах обращения лекарственного средства.
Источник